
Метилмалоновая ацидемия (ацидурия) – генетически гетерогенное наследственное заболевание из группы органических ацидемий, обусловленное блокированием обмена пропионатов на уровне перехода метилмалонил-КоА в сукцинил-КоА и нарушением метаболизма ряда аминокислот (изолейцин, валин, треонин, метионин), жирных кислот с нечетным числом атомов углерода и холестерина. КОД МКБ-10 Е71.1
ЭПИДЕМИОЛОГИЯ
Заболевание встречается в различных популяциях. Частота среди новорожденных в странах Европы 1:48000 - 1:61000; половина случаев представлена В12-резистентными формами. В Российской Федерации точная частота заболевания не определена.
ЭТИОЛОГИЯ, ТИП НАСЛЕДОВАНИЯ
Классическая форма заболевания обусловлена дефицитом метималонил-КоА мутазы (ген MUT) с полным (mut0) или частичным (mut-) отсутствием активности фермента. Кофактором данного фермента является витамин В12, поэтому дефекты метаболизма данного фермента также сопровождаются метилмалоновой ацидурией - изолированной или в сочетании с гомоцистинурией. Локализация гена MUT - 6p21. Формы метилмалоновой ацидурии, связанные с нарушениями метаболизма витамина В12, обусловлены мутациями генов MMAA, MMAB, кодирующих обмен аденозилкобаламина (cblA, cblB формы) или дефицитом метилмалонил-КоА эпимеразы (ген MCEE). Локализация генов 4q31.1-q31.2, 12q24, 2р13.3 соответственно. Крайне редко встречается форма метилмалоной ацидурии, связанная с недостаточностью рецептора транскобаламина и мутацией гена CD320, который картирован на 19p13.2. Формы метилмалоновой ацидурии с гомоцистинурией обусловлены мутациями генов: LMBRD1 – cblF форма, MMADHC – cblD форма и MMACHC- cblC форма. Тип наследования при всех генетических вариантах – аутосомно-рецессивный. Также описаны формы ММА, обусловленные истощением митохондриальной ДНК и связанные с мутациями генов SUCLA2, SUCLG1. Данные заболевания относятся к группе митохондриальных энцефалопатий и тактика их лечения отличается от терапии других форм метилмалоновой ацидурии.
ПАТОГЕНЕЗ
Патогенез заболевания связан с накоплением производных метилмалоновой и пропионовой кислот вследствие блокирования обмена на уровне перехода метилмалонил-КоА в сукцинил-КоА. Предшественниками пропионатов в организме служат аминокислоты изолейцин, валин, треонин и метионин (50% общего количества пропионатов), жирные кислоты с нечетным числом атомов углерода и холестерин (25%); остальная часть пропионатов образуется в кишечнике в результате деятельности эндогенной флоры. Накопление органических кислот (пропионовой, метилмалоновой, метиллимонной кислот и др.) ведет к тяжелому метаболическому кетоацидозу, вторичной гипераммониемии, гиперглицинемии, гипогликемии. Повышенный уровень в крови и высокая почечная экскреция пропионилкарнитина обусловливают истощение запасов карнитина и его вторичный дефицит
КЛАССИФИКАЦИЯ
Выделяют В12-резистентную (около 1/2 случаев) и В12-зависимую формы метилмалоновой ацидурии. В12-резистентная форма характеризуется более ранней манифестацией и тяжелым приступообразным течением. По срокам появления первых признаков болезни различают неонатальную, младенческую и позднюю формы.
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ
В большинстве случаев заболевание имеет кризовое течение. Метаболический криз провоцируется факторами, ведущими к усилению процессов катаболизма: интеркуррентные инфекции, хирургическое вмешательство, приемом больших количеств белка и др. При В12-резистентной форме ММА первые симптомы обычно появляются в возрасте 2 недель – 4 месяцев: упорная рвота, отказ от еды, дегидратация, вялость, сонливость, дыхательные нарушения, задержка психомоторного и физического развития, иногда развиваются инсультоподобные эпизоды. В более старшем возрасте, помимо значительной задержки психоречевого и моторного развития, у детей отмечаются неврологические нарушения, в виде различных эксрапирамидных нарушений (хореоатетоидные и миоклонические гиперкинезы, мышечная дистония, инсультоподобные эпизоды, эпилептические приступы), поражение почек по типу тубулоинтерстициального нефрита с артериальной гипертензией и почечной недостаточностью, эритематозный дерматит, в отдельных случаях - панкреатит и кардиомиопатия. При компьютерном и магнитно-резонансном томографическом исследованиях выявляются характерные нарушения: кортикальная атрофия, расширение желудочков, утолщение борозд, задержка миелинизации, повышение интенсивности сигнала в области базальных ганглиев в T2- взвешенном изображении.
В12-зависимая форма болезни обычно имеет более позднюю манифестацию – после неонатального периода. При всех формах в период метаболического криза у детей отмечаются следующие обменные расстройства: тяжелый метаболический ацидоз, кетоз, гиперглицинемия, гипераммониемия, нейтро- и тромбоцитопения, гиперурикемия, у части больных – гипогликемия. Иногда наблюдается гипергликемия.
В крови повышается уровень пропионилкарнитина (С3), в некоторых случаях метилмалонилкарнитина C4DC и снижается содержание свободного карнитина (С0). В моче значительно повышается концентрация метилмалоновой кислоты, а также 3-гидроксипропионовой, 3-гидрокси-nвалериановой, метиллимонной.
ДИАГНОСТИКА
Диагностика метилмалоновой ацидурии основана на анализе родословной, оценке данных анамнеза, клинических проявлений, результатах анализа уровня аминокислот изолейцина, валина, метионина и треонина в крови, определении содержания в крови пропионилкарнитина (С3) и свободного карнитина (С0), почечной экскреции органических кислот - метилмалоновой, 3-гидроксипропионовой, 3-гидрокси-n-валериановой, метиллимонной, пропионилглицина. Основными методами подтверждения диагноза являются биохимические методы: тандемная масс-спектрометрия (МС/МС), аминокислотный анализ, газовая хроматография-массспектрометрия.
Для подтверждения диагноза и медико-генетического консультирования проводится молекулярно-генетическое исследование.
Обследованию на метилмалоновую ацидурию подлежат следующие группы детей:
- дети любого возраста из семей, имеющих больных с данным заболеванием (в первую очередь, братья и сестры больного);
- дети первых недель и месяцев жизни, у которых после некоторого периода (иногда очень короткого, в течение нескольких суток) удовлетворительного состояния появились рвота, отказ от еды, летаргия, гипотония, судороги, кома, метаболический ацидоз, кетонурия;
- дети любого возраста с повторными приступами рвоты, вялости, сонливости, гипотонии, кетоацидоза;
- дети, отстающие в психомоторном развитии, с эпилепсий, нарушением мышечного тонуса
Для установления диагноза метилмалоновой ацидурии у пациентов с клиническими симптомами заболевания специфическими диагностическими тестами следует считать (сила А по Оксфордской шкале):
- количественное определение пропионилкарнитина (С3), свободного карнитина (С0), глицина в крови;
-количественное определение метилмалоновой, 3-гидроксипропионовой, 3-гидрокси-n-валериановой, метиллимонной кислот в моче;
- выявление мутаций в генах MUT, MMAA, MMAB, MCEE.Биохимические методы диагностики Методом тандемной масс-спектрометрии (МС/МС) в плазме крови, в пятнах высушенной крови выявляют концентрацию аминокислот и ацилкарнитинов. При метилмалоновой ацидурии наблюдается повышение концентрации С3 (пропионилкарнитина) и С4DC (метилмалонилкарнитина).
Необходимо с большой осторожностью относится к интерпретации данных показателей, которые являются возраст-зависимыми. В ряде случаев, концентрация С3 может быть в пределах нормы, а повышается соотношение С3/С2.
Аминокислотный анализ Уровень изолейцина, валина, метионина и треонина в крови у многих пациентов может быть в пределах нормы, что не позволяет на основании анализа аминокислот подтвердить или исключить заболевание. У многих пациентов с метилмалоновой ацидурией в крови и моче может наблюдаться повышение уровня глицина. Однако, это не является специфичным и может наблюдаться при других наследственных нарушениях обмена веществ. Наиболее важным для дифференциальной диагностики служит определение уровня гомоцистеина в крови, концентрации гомоцистина в моче.
Методом газовой хроматографии масс-спектрометрии в моче пациентов выявляют повышение концентрации метилмалоновой кислоты, а также ряда ее производных (табл. 1). По уровню метилмалоновой кислоты в моче невозможно точно установить форму заболевания, однако наиболее высокие концентрации обычно встречаются при метилмалоновой ацидурии, обусловленной мутациями в генах MUT и MMAB.
Таблица 1. Концентрация метаболитов в моче при метилмалоновой ацидурии
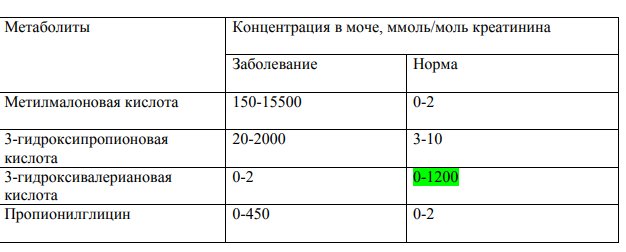
Также в разной степени могут повышаться другие органические кислоты и их производные: 2-метилглутаконовая, 3-кето-2-метилмасляная, метиллимонная, 3-гидроксибутират, лактат, пируват. На основании анализа органических кислот мочи возможно проведение дифференциальной диагностики с другими формами органических ацидурий. Наиболее близкой по клиническим проявлениям и по изменениям спектра ацилкарнитинов является пропионовая ацидурия, при которой не наблюдается повышение концентрации метилмалоновой кислоты в моче, а присутствуют только метаболиты пропионовой кислоты.
Можно рекомендовать после получения результатов тандемной массспектрометрии проведение простого качественного теста на присутствие метилмалоновой кислоты в моче.
На культуре кожных фибробластов возможно осуществление нагрузочных тестов, которые позволяют более точно установить форму метилмалоновой ацидурии. Поскольку эти исследования сопряжены с применением инвазивной технологии (забор культуры фибробластов), связаны с техническими трудностями (исследования проводятся лишь в единичных лабораториях мира) и работой с мечеными изотопами, с целью установления формы заболевания предпочтительным являются молекулярногенетические исследования.
Молекулярно-генетические методы диагностики
С помощью стандартных молекулярно-генетических методов проводят исследование генов, ответственных за развитие метилмалоновой ацидурии. Проведение ДНК-диагностики строго показано для пренатальной или преимплантационной диагностики. В ряде случаев, установление первичного молекулярно-генетического дефекта позволяет скорректировать тактику ведения пациента. Лечение пациентов с метилмалоновой ацидурией должно начинаться незамедлительно после подтверждения диагноза биохимическими методами.
ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
Дифференциальная диагностика проводится с гипоксическими поражениями нервной системы, внутриутробными инфекциями, поствакцинальными осложнениями, сахарным диабетом (при выявлении гипергликемии), наследственными нарушениями обмена веществ, в частности с другими формами органических ацидурий и с дефектами цикла синтеза мочевины.
ЛЕЧЕНИЕ
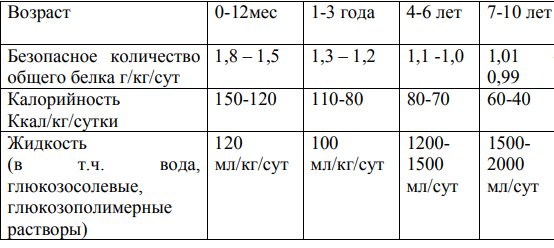
Стратегия лечения больных заключается в снижении образования пропионатов, предупреждении развития кетоацидоза, гипераммониемии, токсического поражения тканей головного мозга и внутренних органов с обеспечением процессов анаболизма, нормального роста и нутритивного статуса детей. Тактика лечения основана на следующих принципах. При подозрении на наследственное нарушение обмена аминокислот и органических кислот начинать диетическое лечение необходимо сразу после взятия анализов, не дожидаясь их результатов. До получения результатов, подтверждающих метаболическое заболевание, следует ограничить поступление белка: для детей первого полугодия жизни – грудное вскармливание или вскармливание детскими молочными смесями с низким содержанием белка (1,2 -1,3 г/100 мл восстановленного продукта). Для детей старше 6 месяцев - исключение высокобелковых продуктов (мясо, творог, рыба) из рациона. После подтверждения диагноза тактика лечения основана на следующих принципах:
- ограничение поступления изолейцина, валина, треонина и метионина с пищей до минимальной потребности;
- обеспечение физиологической потребности в других аминокислотах и необходимых нутриентах для предупреждения их недостаточности и поддержания анаболизма;
- ограничение потребления жирных кислот с нечетным числом атомов углерода и холестерина;
- назначение левокарнитина и глицина для усиления связывания токсичного пропионил-радикала;
- коррекция вторичной карнитиновой недостаточности;
- кофакторная терапия витамином В12;
- исключение голодания, предупреждение активации процессов катаболизма;
- контролирование кислотно-основного состояния крови, предотвращение развития ацидоза, поддержание водного баланса;
- усиление терапии в период метаболического криза.
Основными компонентами комплекса лечения больных служат малобелковая диетотерапия, препараты левокарнитина и витамин В12 при В12- зависимой форме. Терапию дополняют назначением глицина, антибактериальных препаратов, других витаминов группы В, по показаниям антиконвульсантов, симптоматических средств.
Основные принципы диетического лечения:
- Строгое ограничение белка натуральных пищевых продуктов с целью сведения к необходимому минимуму поступление в организм аминокислот метионина, треонина, валина, изолейцина. Для детей первого полугодия жизни ограничение распространяется на материнское молоко или детские молочные или соевые смеси, для детей второго полугодия жизни – запрет на введение в качестве прикорма высокобелковых продуктов (мяса, рыбы, творога, яиц, молочных продуктов, бобовых и т.д.), для пациентов старше одного года – строгие ограничения в использовании высокобелковых продуктов.
- Обязательная компенсация дефицита белка за счет специализированных смесей на основе аминокислот, соответствующих возрастным потребностям ребенка в основных пищевых веществах и энергии, но не содержащих метионин, треонин, валин, изолейцин. В начале лечения и в период метаболических кризов в течение 24 – 72 часов (не более!) использовать только специализированную смесь аминокислот.
- С целью поддержания процессов анаболизма и предотвращения развития процессов катаболизма обеспечение достаточной энергетической ценности рациона в основном за счет углеводов.
- Ограничение квоты жиров до 50-60% от возрастных суточных потребностей во избежание накопления пропионовой кислоты - продукта β-окисления жирных кислот с нечетной длиной цепи. В качестве источника жира предпочтительнее использовать растительные масла, содержащие полиненасыщенные жирные кислоты (рапсовое, льняное, оливковое, подсолнечное и др.).
- Обеспечение пациента достаточным количеством жидкости.
- Дробное кормление без длительных ночных перерывов во избежание голодания, особенно у детей грудного и раннего возраста.
- Психологическая поддержка и обучение родителей правилам организации помощи и диетотерапии в межприступный период и в период угрозы метаболического криза.
- У родителей ребенка и при ребенке всегда должна быть памятка с указанием неотложных мероприятий в период угрозы и развития метаболического криза.
Лечение детей вне периода метаболического криза
Диетотерапия в межприступный период:
- В межприступный период диетотерапия осуществляется в соответствии с вышеописанными принципами.
- Общий белок лечебного рациона рассчитывается исходя из возрастных потребностей ребенка, а также с учетом толерантности пациента к белку в зависимости от формы заболевания и тяжести его течения.
- Квота белка натуральных продуктов назначается с учетом минимальной потребности в патогенетически значимых аминокислотах – метионине, треонине, валине, изолейцине (табл. 4).
- Рекомендуется пользоваться одними справочными материалами по химическому составу продуктов, так как в разных справочниках данные могут отличаться.
- С целью компенсации дефицита белка используются специализированные продукты на основе аминокислот без метионина, треонина, валина, изолейцина (табл. 5).
Таблица 2. Среднесуточные нормы потребностей в основных пищевых веществах и энергии для детей первого года жизни (на кг массы тела)
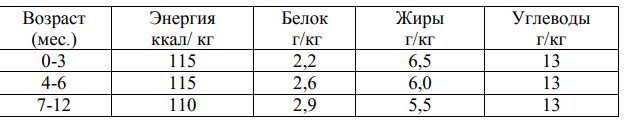
Таблица 3. Нормы физиологической потребности в основных пищевых веществах и энергии для здоровых детей старше года*
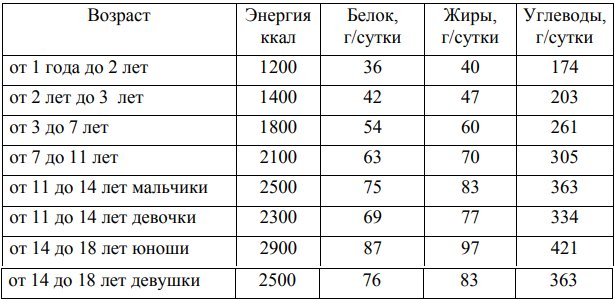
* -для детей с метилмалоновой ацидурией старше года потребление белка по сравнению с указанными в таблице уменьшается на 10-25% в зависимости от формы заболевания, тяжести состояния и нутритивного статуса ребенка.
Таблица 4. Ориентировочная потребность в метионине, треонине, валине, изолейцине у больных метилмалоновой ацидурией в зависимости от возраста
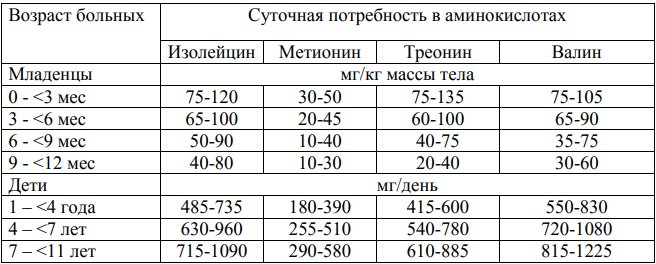
Таблица 5. Специализированные продукты на основе аминокислот без метионина, треонина, валина, изолейцина.

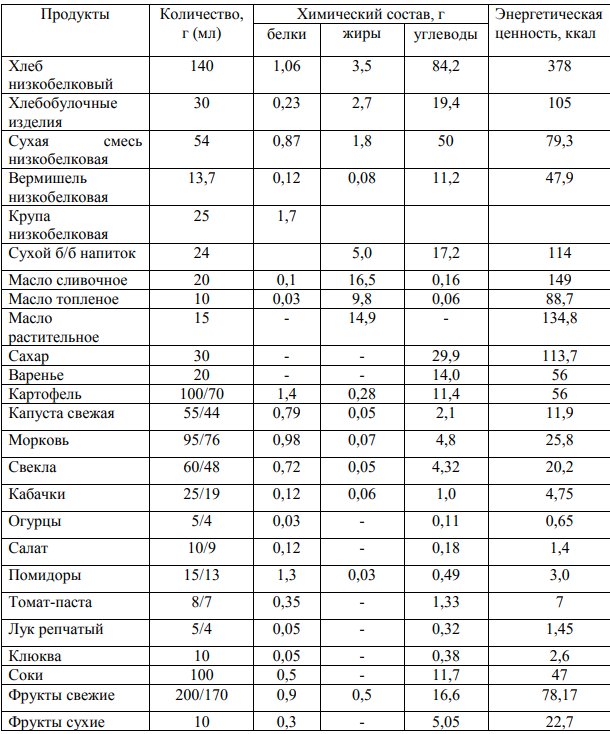
В таблице 6 представлен суточный набор продуктов для детей дошкольного возраста с метилмалоновой ацидемией
Таблица 6. Примерный суточный набор продуктов, его химический состав и энергетическая ценность для детей дошкольного возраста с метилмалоновой ацидемией
Кофакторная терапия.
При установлении диагноза метилмалоновой ацидурии необходимо провести пробное лечение витамином В12 для определения формы заболевания - В12-резистентная или В12-зависимая. С этой целью назначают короткий курс (8-10 дней) витамина В12 в дозе 1 мг/сут под контролем уровня пропионилкарнитина в крови и экскреции метилмалоновой кислоты. Снижение этих показателей свидетельствует о В12- зависимой форме болезни. При получении сомнительных результатов рекомендуется продолжить исследование, вдвое увеличив дозу препарата. Детям с установленной В12-зависимой формой метилмалоновой ацидемии назначают длительное лечение витамином В12 в средней дозе 1-5 мг/сут (в отдельных случаях до 15 мг/сут). Более эффективно применение активных форм витамина – гидроксикобаламин, метилкобаламин, аденозилкобаламин (кобамамид). Как правило, пациентам дополнительно назначают диетическое лечение с умеренным ограничением белка натуральных продуктов (напр., до 1,2 г/кг массы).
Терапия левокарнитином.
С целью усиления связывания пропионового радикала и ликвидации карнитиновой недостаточности больным назначают большие дозы карнитина. Рекомендуется использовать только L-формы карнитина (элькар 30%, карнитен). Дети раннего возраста получают карнитин из расчета 100-150 мг/кг массы тела в сутки за 2-3 приема, дети более старшего возраста – 60-80 мг/кг/сут. Суточная доза не должна превышать 3 г. Терапия проводится непрерывно.
Антибактериальная терапия.
Учитывая, что около 1/4 количества пропионатов образуются в кишечнике под влиянием функционирования местной бактериальной флоры, дополнительным способом коррекции обмена является антибактериальная терапия. С целью подавления активности кишечной флоры больным показаны курсы антибактериальных препаратов. Назначают ампициллин в возрастной дозе в течение 8-10 дней или метронидазол в дозе 10-15 мг/кг/сут в течение 10-14 дней; через 3-4 недели курс повторяют.
Другие используемые лекарственные средства.
Дополнительно в комплексе лечения детей применяют глицин, который, подобно карнитину, обладает способностью конъюгировать производные органических кислот. Суточная доза составляет 0,3-0,6 г/сут в течение 4-6 мес. Назначают витамины группы В в возрастных дозах. По показаниям используют антиконвульсанты; ноотропные препараты назначают с осторожностью во избежание провокации эпилептических приступов.
Лечение детей в период метаболического криза
Метаболические кризы, ведущие к критическим, угрожаемым жизни состояниям, при метилмалоновой ацидурии проявляются в виде остро возникающей энцефалопатии и приступов рвоты, возможно развитие судорог. Кризы обычно провоцируются разными неблагоприятными факторами, которые обусловливают усиление процессов клеточного катаболизма с образованием токсичных пропионовых производных:
- прием белка и липидов в количестве, превышающем толерантность организма больного ребенка;
- недоедание, низкая калорийность рациона;
- интеркуррентные респираторные или желудочно-кишечные инфекционные заболевания;
- вакцинация;
- физическая или психо-эмоциональная нагрузка.
Ранними признаками метаболического криза служат вялость, сонливость или раздражительность, отказ от еды, рвота. Неврологические расстройства прогрессируют вплоть до ступора или комы, обусловливая высокую летальность. Тяжесть состояния, главным образом, определяется выраженным метаболическим кетоацидозом. Кроме того, могут иметь место гипогликемия (в некоторых случаях гипергликемия), гипераммониемия, обезвоживание. Состояние метаболического криза, особенно у детей раннего возраста, является показанием для госпитализации и основанием для проведения интенсивной терапии.
При угрозе или в случае развития метаболического криза лечение должно начинаться незамедлительно. Лечебные мероприятия направлены на прекращение образования и накопления токсичных органических соединений и выведение их из организма. Тактика лечения детей в период криза включает коррекцию диетотерапии, активацию связывания накапливающихся органических кислот путем увеличения дозы левокарнитина, коррекцию метаболического ацидоза, гипераммониемии и водно-электролитных нарушений, дополнительное введение глюкозы для энергетической поддержки и уменьшения интенсивности процессов катаболизма.
Диетотерапия детей в период метаболического криза:
- Перевести ребенка на питание исключительно смесью аминокислот без изолейцина, метионина, треонина и валина, но не более чем на 24-72 часа. Соблюдать режим дробных и частых кормлений с промежутками между кормлениями 2-3 часа, возможно непрерывное капельное вскармливание через инфузомат. При наличии срыгиваний, рвоты, отказа от еды – кормление через назогастральный зонд или гастростому.
- Количество общего потребляемого белка должно быть не ниже безопасного уровня (табл. 8).
- Обеспечить высококалорийное питание (табл. 8) за счет использования 5-10% глюкозы, глюкозополимерных растворов (мальтодекстрин) в дополнение к смеси на основе аминокислот; расчет производится исходя из калорийности 1 г углеводов = 4 ккал, 1 г мальтодекстрина приравнивается к 1 г углеводов. Возможно парентеральное введение углеводов (5%-10% глюкозы), а также липидов до 1 г/кг/сутки.
- Через 24-72 часа от начала лечения постепенно вводят продукты, содержащие натуральный белок, из расчета 1/4 необходимого суточного объема – в первый день, 1/2 - на 2-3-й день, 3/4- 3-4-й день, далее в полном объеме. Источником натурального белка для детей первых шести месяцев жизни является материнское молоко/детская молочная смесь, для детей второго полугодия жизни – также низкобелковые продукты прикорма, для детей старше года - низкобелковые натуральные продукты (крупы, овощи, фрукты, растительные масла) и специализированные продукты на основе крахмалов.
- С целью поддержания соответствующей энергетической ценности рациона продолжают использовать мальтодекстрин, а также низкобелковые продукты на основе крахмала.
- В период выхода из метаболического криза пища должна иметь щадящую кулинарную обработку.
- Последующее увеличение квоты натурального белка в рационе проводят по мере стабилизации метаболических нарушений, в соответствии с нутритивным статусом ребенка и его двигательной активностью
Таблица 8. Диетические рекомендации в период метаболического криза
Активация связывания накапливающихся органических кислот. Суточную дозу карнитина резко увеличивают до 300-400 мг/кг. Предпочтительно внутривенное или внутримышечное введение препарата. Цель терапии – поддержание содержания свободного карнитина в крови выше нормальных значений для усиления связывания накапливающихся пропионил-радикалов и их выведения из организма в виде пропионилкарнитина.
Коррекция метаболического ацидоза осуществляется внутривенным введением щелочных растворов гидрокарбоната натрия, трисоля или (трисбуфера)трисамина (ТНАМ). Гидрокарбонат натрия применяется в виде 8,4% и 4,2% раствора для удобства перерасчета на ммоль NаНСО. Его дозировка (ммоль) определяется по формуле: (-ВЕ) Х масса тела (кг) Х 0,3. Кроме того, больным рекомендуется щелочное питье – раствор соды из расчета ?-1 чайная ложка на 200 мл воды, щелочные минеральные воды. Можно использовать введение соды в виде ректальных свечей. Регулярно (каждые 6-12 часов в зависимости от тяжести состояния) контролируют показатели кислотно-основного состояния крови.
Коррекция водно-электролитных нарушений, гипераммониемии и энергетической недостаточности. Для устранения гипогидратации назначают внутривенное введение физиологического раствора, используя следующий расчет в зависимости от массы ребенка: 100 мл/кг/сут (масса <10 кг.); 1000 мл + 50 мл/кг на последующий килограмм после 10 кг (масса 10-20кг); 1500 мл + 20 мл/кг на последующий килограмм после 20 кг (масса >20 кг). В случае тяжелой интоксикации для лучшей элиминации накапливающихся пропионатов и метилмалоновой кислоты рекомендуется использовать метод форсированного диуреза с дополнительным введением жидкости и назначением фуросемида (лазикса) в дозе 1-3 мг/кг 1-2 раза в сутки (с интервалом не менее 6-8 час.).
С целью устранения энергетического дефицита и снижения уровня аммиака в крови проводят внутривенное введение 10%-20% раствора глюкозы из расчета 20 мл/кг с инсулином (1 Ед./8 г глюкозы). После двухчасового введения рекомендуется контроль содержания лактата и глюкозы в крови; допустимо поддерживать уровень глюкозы выше 3,3 ммоль/л у новорожденных и 5,5 ммоль/л – у старших детей. При уровне аммиака в крови выше 200 мкмоль/л дополнительно для стимуляции синтеза мочевины показано введение аргинина (250-300 мг/кг) или цитруллина (350 мг/кг). Назначают фолиевую кислоту 0,1 мг/кг/сут, пиридоксин 5 мг/сут. Осуществляют обязательный контроль уровня натрия и калия в крови. Пациентам в состоянии острого метаболического криза, при отсутствии быстрой положительной реакции на интенсивную терапию, сохраняющемся кетоацидозе и гипераммониемии, для более эффективного выведения токсичных метаболитов проводят перитонеальный диализ или гемодиализ.
Контроль терапии
В процессе комплексного лечения осуществляют контроль показателей клинического анализа крови, уровня гемоглобина, общего белка, альбумина, глюкозы, сывороточного железа, электролитов, лактата, аминокислот, свободного карнитина и пропионилкарнитина. Контролируют параметры кислотно-основного состояния крови. Проводят определение почечной экскреции метилмалоновой кислоты. Кратность проведения анализов зависит от состояния ребенка, но в период инфекционных заболеваний, метаболического криза определение аминокислот и карнитинов осуществляют не реже 1 раза в 7-10 дней, КЩС - ежедневно до стабилизации показателей.
Уровни метионина, треонина, валина, изолейцина в крови в результате лечения должны быть приближены к референсным возрастным значениям (табл. 9). Особенно важно обеспечивать нормальный сывороточный уровень аминокислот с разветвленной углеродной цепью. Так, недостаточное потребление белка и дефицит изолейцина являются причиной развития тяжелого акродерматита и служат основанием для дополнительного введения в рацион питания L-изолейцина в дозе 50-100 мг/сут в зависимости от степени дефицита.
Таблица 9. Референсные значения изолейцина, метионина, треонина и валина в плазме крови детей

Показатель свободного карнитина поддерживают на высоком уровне, превышающем границы, определенные для здоровых детей. Это обеспечивает детоксикационную функцию карнитина, улучшает связывание и выведение из организма токсичных пропионовых производных. При использовании высоких доз возможно развитие эффектов в виде тошноты и неприятного запаха. Указанные явления проходят после снижения дозы. Осуществляют контроль нутритивного статуса с коррекцией питания в зависимости от состояния ребенка и его толерантности к белку. Контроль фактического питания (химического состава рациона) для предупреждения развития дефицитных состояний проводят у детей в реанимационном периоде – ежедневно, у детей первого года жизни - не реже 1 раза в 7-10 дней, у пациентов старше 1 года– 1 раз в 1-3 месяца.
Лабораторными предвестниками развития кетоацидотического криза служат тенденция к снижению pH крови, дефициту оснований, уменьшение содержания свободного карнитина, нарастание уровня пропионилкарнитина в крови, почечной экскреции метилмалоновой кислоты и кетоновых тел. Родители должны быть обучены правилам организации терапии в межприступный период и в период угрозы метаболического криза. У родителей ребенка и при ребенке всегда должна быть памятка с указанием неотложных мероприятий в период начинающегося метаболического криза. Кроме того, больные нуждаются в наблюдении нефролога, кардиолога, гастроэнтеролога в связи с риском тубулоинтерстициального нефрита, кардиомиопатии, панкреатита - контроль за клиническими анализами мочи (1 раз в 6 мес.), ЭхоКГ, УЗИ внутренних органов (1 раз в год).
ХИРУРГИЧЕСКОЕ ЛЕЧЕНИЕ
В ряде случаев (при тяжелом поражении почек, плохо корректируемых метаболических нарушениях) возникает необходимость решать вопрос о трансплантации печени и почек.
ПРОГНОЗ
Прогноз состояния и уровня психического развития больных зависит от тяжести заболевания (В12-резистентная форма mut0 отличается более ранней манифестацией, большей степенью тяжести болезни и нестабильностью течения), вовлечения в патологический процесс внутренних органов (почки, сердце, поджелудочная железа), а также сроков начала специализированной терапии и качества лечения, способного предупредить приступы метаболической декомпенсации.
Пренатальная диагностика
Пренатальная диагностики возможна с помощью молекулярногенетического исследования биоптата хориона с выявлением мутации соответствующего гена.
Источник: МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РФ, Федеральные клинические рекомендации по диагностике и лечению метилмалоновой ацидемии, Москва 2013


 Нет ли у вас панкреатита?
Нет ли у вас панкреатита?

.jpeg)




